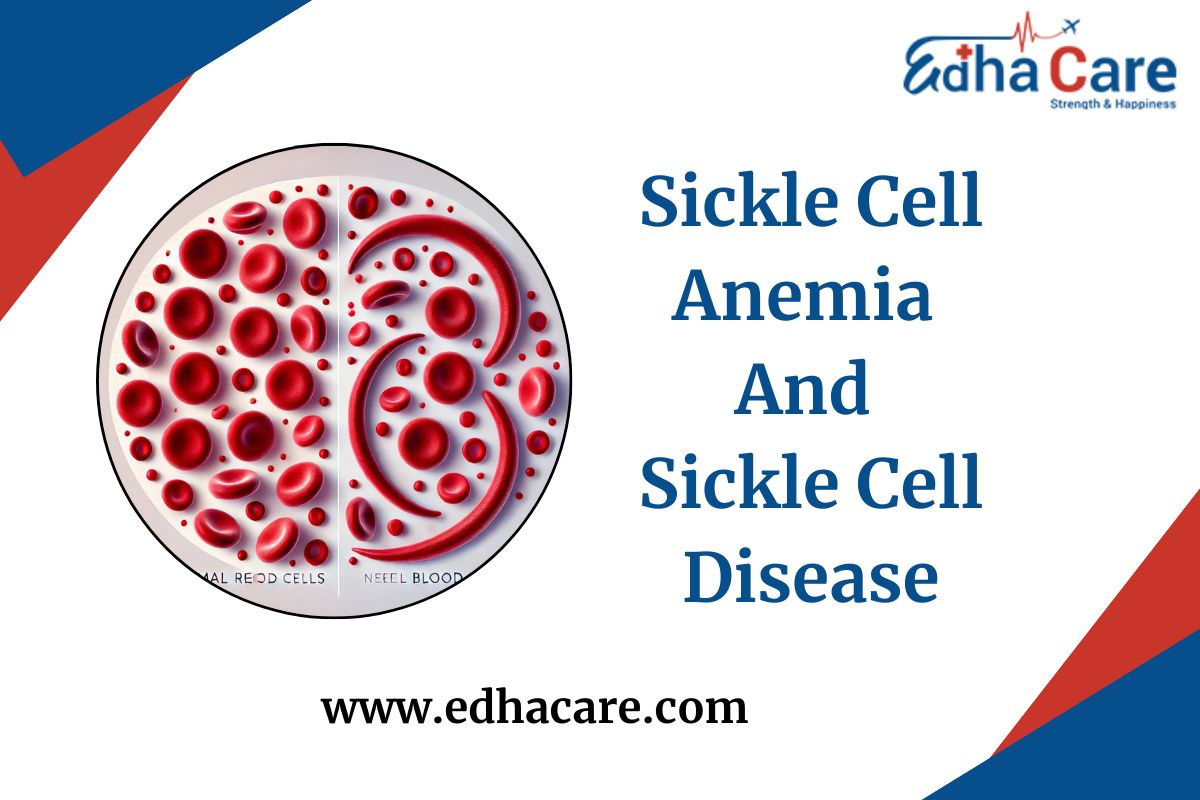
Sickle cell disease and sickle cell anemia are frequently confused and utilized interchangeably. But there is a subtle difference between the two. SCD is a collection of inherited disorders of the red blood cells. Sickle cell anemia usually occurs as the most common and severe form of SCD. The present blog post will delve into these conditions and will explore these causes, symptoms, diagnosis, management, and what type of research is going on concerning better treatments and a potential cure.
What is Sickle Cell Disease and its Cause?
Genetically, SCD is caused by a mutated gene from the body telling it how to mold the hemoglobin component of red blood cells. It declares the red blood cells as being sickle-shaped by forcing them to be rigid, curving them into a crescent shape instead of their normal rounded, flexible form.
The sickle-shaped cells block off blood flow by getting caught in narrow blood vessels. As a result of pain and other health complications. Complications of the disease include anemia, pain crises, infections, stroke, and damage to the organs.
What is Sickle Cell Anemia and its Cause?
More specifically sickle cell anemia describes a person who has inherited two copies of the mutated gene, one from each parent. This produces a considerable amount of hemoglobin S and hence generally increases the chance of symptoms. There are other forms of SCD such as sickle cell trait (inheritance of one copy of the mutated gene with one normal gene). And other compound heterozygous conditions in which the patient has one sickle cell gene and another abnormal hemoglobin gene (for example: hemoglobin C, beta thalassemia). Most of these other variants may have milder symptoms or no symptoms at all.
Symptoms of Sickle Cell Anemia and Disease:
These signs for SCD will be very personal and differ from one patient to the next, ranging from mild to severe symptoms. Some individuals with SCD have almost no symptoms, while others may present with very frequent and debilitating episodes. The common signs of sickle cell disease include:
- Pain: Pain is the cardinal symptom of sickle cell disease. This occurs when the sickle cells block the circulation, damaging the tissue and making it inflamed. The pain is also called a vaso-occlusive crisis. It can occur in any part of the body and lasts from hours to days.
- Anemia: Sickle cells live a shorter period as compared to normal red blood cells so, cause less amount of red blood cells, thus, anemia occurs. The symptoms include fatigue, weakness, shortness of breath, and dizziness.
- Jaundice: The breakdown of sickle blood cells releases bilirubin into the bloodstream. This yellow pigment causes jaundice characterized by yellowness in the skin and eyes.
- Frequent Infections: Sickle cell disease damages the spleen, which fights infections. All these make a sickler susceptible to infections.
- Delayed Growth and Development: Children having Sickle-cell Anemia grow and develop at a pace much slower than those without Sickle-cell Anemia.
- Acute chest syndrome: It is a serious complication of SCD, characterized by chest pain, fever, cough difficulty breathing, usually triggered by a lung infection.
- Stroke: blood flow blockage by sickle cells can result in a stroke due to the subsequent brain tissue damage.
- Organ Damage: Repeated blocked blood flow leads to gradual damage to organs like the kidneys, lungs, heart, and eyes.
- Leg ulcer: What ultimately occurs is that painful ulcers develop as a result of poor circulation in the legs.
- Vision Problems: sickle cell disease can also affect the blood vessels in the eyes and further lead to blindness.
Diagnosis of Sickle Cell Anemia and Sickle Cell Disease:
Both Sickle cell anemia and Sickle cell disease are diagnosed in the same way.
SCD and SCA are usually diagnosed through a blood test called hemoglobin electrophoresis. In this test, the various types of hemoglobin in the blood are measured and the presence of hemoglobin S is determined. Newborn screening programs routinely screen for SCD to allow early diagnosis and intervention. Genetic testing may be done to confirm the diagnosis and to determine if an individual is a carrier of the sickle cell trait.
Management and Treatment of Sickle Cell Disease & Anemia:
Management of SCD aims at the prevention and treatment of complications and enhancing life quality. The medications used include:
- Pain Management: Pain events are usually treated using pain medications, including over-the-counter pain relievers and in severe cases stronger opioids. Hydration also constitutes a serious aspect during episodes of pain.
- Hydroxyurea: This drug decreases the occurrence of pain episodes and other complications because it stimulates the production of fetal hemoglobin-the type of hemoglobin that does not sickle.
- Blood transfusions: Blood transfusions can alleviate anemia and lower the chances of stroke.
- Bone Marrow Transplant (Hematopoietic Stem Cell Transplantation): This is the only curative medicine for SCD; however, it is a difficult and hazardous kind of procedure. This procedure is mainly reserved for children having extreme types of SCD.
- Gene Therapy: Research is still being conducted; however, gene therapy has a very promising new therapeutic option in a possible cure for the disease, targeting the faulty gene within sickle cell disease.
- Drugs to Decrease Sickle Cell Adhesion: More modern drugs are being produced to stop sickle cells from sticking along the walls of blood vessels, thus reducing the incidences of vaso-occlusive crises.
- Support Care: This includes management of anemia, prevention of infections, and damage caused to organs.
Living with Sickle Cell Disease:
Living with SCD can be challenging; however, given the proper management and support, SCD-stricken people can lead normal lives. Importantly, working alongside a close-knit healthcare team including hematologists, pain specialists, and other specialists is necessary and crucial. Lifestyle considerations (healthy living with regular exercise, a balanced diet, no smoking, and recreational alcohol use). It can also improve the overall health and well-being of an individual.
Research in Progress:
Research into sickle cell anemia and disease is ongoing. This developing new and improved therapeutic strategies such as gene therapy and other targeted therapies. This also targets defining the understanding of the mechanisms of sickle cell disease. Finally, they try to find new targets for complications prevention.
Conclusion:
Sickle cell anemia is the commonest common type of sickle cell disease. Sickle cell disease is a difficult and complex condition. However, advances in the diagnosis and treatment of SCD have dramatically enhanced the quality of life for those affected by the disease. Continued research and focused care are optimistic for improved outcomes in the future.
If you or someone you know has been struck by SCD. It is very important to seek medical help from a health professional who has enough knowledge in treating such a condition. Through early diagnosis, comprehensive management, and persistent research. Consulting EdhaCare can be the first step toward a healthier life.